FAQs
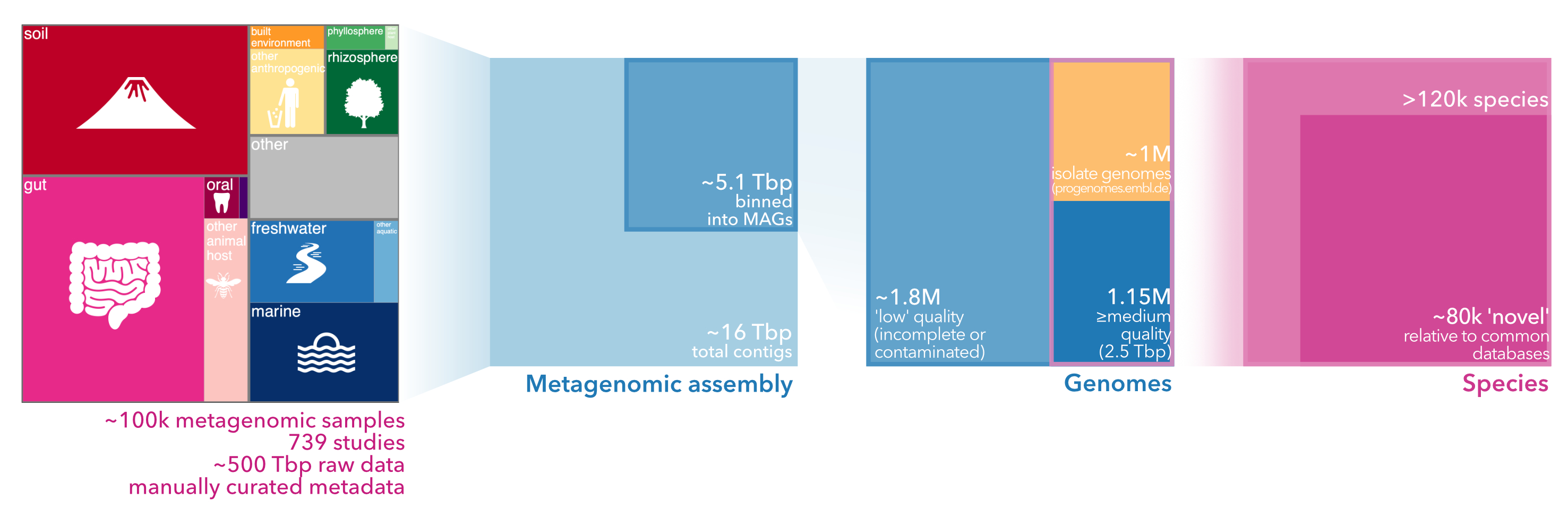
SPIRE is short for Searchable Planetary-scale mIcrobiome REsource: a one stop shop for microbial data, integrated and consistently processed across habitats and phylogeny, at global scales. SPIRE holds five main types of data, derived from ~100k metagenomic samples in 739 studies encompassing ~500Tbp of publicly available raw metagenomes:
-
manually curated contextual data, including annotations against a custom microntology (See FAQ: What is microntology?)
-
~16Tbp worth of metagenomic assemblies (contigs)
-
~35 billion metagenomic open reading frames (ORFs) called from these contigs, functionally annotated in various ways 1.16 million metagenome-assembled genomes (MAGs) of medium or high quality, co-clustered with isolate genomes into over 100 thousand species-level clusters
-
a custom mOTU database mOTUs to allow taxonomic profiling against these species-level clusters, as well as pre-computed taxonomic profiles (coming soon...)
The aim of SPIRE is to provide an integrated view of microbial diversity at various taxonomic granularities, at genome and at gene level, across known microbial habitats.
The website is organized around two major modes of data browsing: by environment/habitat and by taxonomy.
-
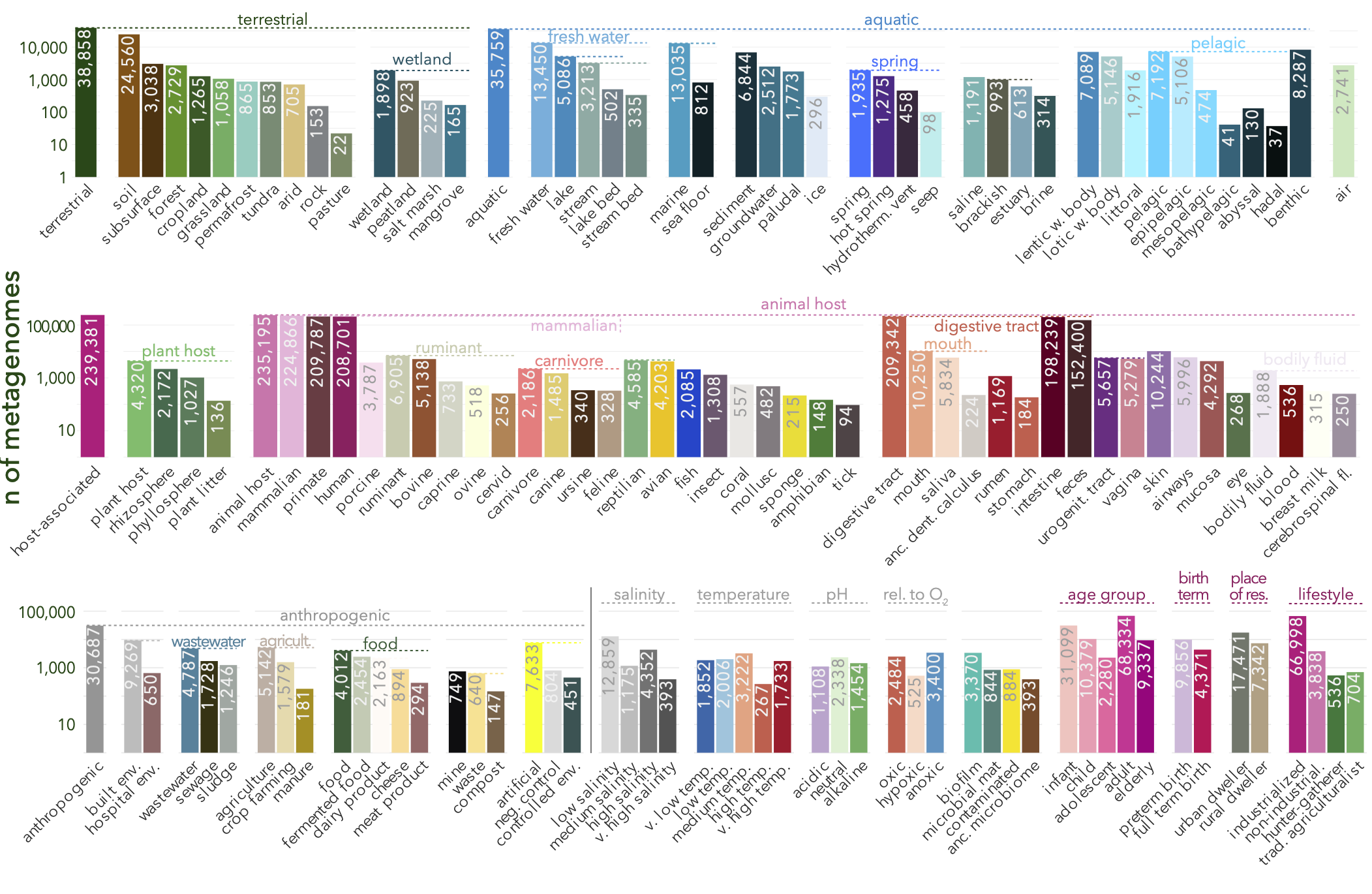
Browsing by environments allows you to filter samples based on combinations of annotated terms and categories using a lightweight microntology (See FAQ: What is microntology?). You can click on individual tags at various hierarchical levels (e.g., 'host-associated => animal host => mammalian host => human host') to subselect samples annotated with these terms. Clicking multiple tags will further subselect to samples carrying the selected combination of terms; you can add/remove tags freely to explore various combinations. The website will display a list of samples meeting the selected criteria and a list of studies containing such samples.
-
Browsing by taxonomy allows you to navigate genomes and species-level clusters across taxonomic clades. You can either search for taxa or click through hierarchical levels (domain, phylum, class, order, family, genus, species, SPIRE species-level cluster) to subset lists of genomes and/or SPIRE genome clusters.
SPIRE holds data on 739 studies, defined as consistent and associated sets of metagenomic samples generated. In most cases, a study in SPIRE will correspond to one project accession in INSCD databases (i.e., European Nucleotide Archive, Sequence Read Archive, DNA Data Bank of Japan). However, there are several exceptions to this general rule:
-
Large consortium projects that generate or accumulate data over longer periods and under multiple separate project accessions are summarized into individual studies; this e.g. applies to the Human Microbiome Project, TARA Oceans or MetaSUB.
-
Data generated by the Joint Genome Institute is usually submitted to INSCD databases under a 'one project accession per biosample' model; as a result, a multi-sample study will correspond to multiple distinct project accessions on ENA or SRA. In SPIRE, we summarize samples under a common umbrella study which is either defined based on a publication (see below) or on JGI-submitted metadata.
-
SPIRE contains a small set of samples that are not directly sourced from INSCD databases; in such cases, studies are defined based on the underlying projects
All studies are assigned human-readable names in SPIRE, usually based on matched primary publications. Names generally follow the pattern "[lastname-of-firstauthor][publication-year][descriptive-tag]" where the "[descriptive-tag]" provide a human-readable keyword on the publication or dataset (e.g., sampled biome or country/region of sampled individuals, studied disease, etc). We invest considerable effort into matching datasets to primary publications, but for a subset of studies, we were unable to do so. Such "studies" retain anonymous yet descriptive tags; in particular, datasets derived from JGI submissions often carry tags of the form "JGI_[descriptive-tag]". If you notice an error in how we matched publications, or if you can point us to a primary publication we missed, please provide feedback via LINK.
SPIRE holds data on 99,146 shotgun metagenomic samples. In most cases, a "sample" in SPIRE corresponds to a biosample in INSDC databases (ENA, SRA, DDBJ). Data processing is performed on the combination of all metagenomic data runs submitted for the same biosample (i.e., runs for the same sample are pooled). However, we curate samples to rectify several types of error in the underlying data: in some projects on INSDC, individual biosamples are erroneously submitted under the same biosample accession, but as distinct experiments or runs; such cases are disambiguated in SPIRE in some projects, the same sample is submitted to INSDC multiple times under different accessions; where possible, we correct this as well
As a consequence, a minority of SPIRE samples do not map 1:1 to biosample accessions in INSDC databases. Moreover, a small subset of data is not directly sourced from INSDC in the first place. The vast majority of samples in SPIRE are shotgun metagenomes directly from environmental samples. We exclude several types of 'artificial' datasets, such as e.g. mock communities or gnotobiotic mouse metagenomes, and specifically flag others (such as e.g. microcosm experiments). A subset of samples is derived from cell sorting experiments, i.e. enriched for certain types of cells and not representative of the initial underlying community. These are likewise flagged, but are included as they contribute significant diversity to the phylogenetic space covered in SPIRE.
Contextual data for each sample in SPIRE is sourced (i) via annotation fields in ENA, (ii) via JGI IMG/M metadata tables where applicable and (iii) directly from matched publications. Information is consolidated into common fields (e.g., latitude and longitude data is manually harmonized across different submitted formats). All samples are manually annotated against a newly developed 'microntology' (See FAQ: "What is microntology?"), a shallow and lightweight ontology of 92 terms to describe microbial habitats and lifestyles, crosslinked to terms in established resources such as the EnvO or UBERON ontologies.
Microntology is a newly developed shallow and lightweight ontology to describe microbial habitats and lifestyles, crosslinked to terms in other established ontologies. Microntology terms in SPIRE are annotated using a 'multiple tag' system, meaning that each sample is described using a combination of concurrent microntology tags, rather than one specific term in a (deep) hierarchy, allowing an annotation with increased flexibility, yet compatibility to established ontologies. (see PREPRINT for more details) The full list of microntology terms and additional information is available via the Downloads page.
The various data types in SPIRE can be accessed and downloaded in different ways. The Downloads page provides bulk summary tables describing all MAGs and all species clusters in SPIRE, all metagenomic gene calls, and the newly generated custom database for mOTU profiling. Metagenomic assemblies and MAGs can moreover be accessed on a per-sample and a per-genome basis, or in bulk on a per-study basis. We are currently in the process of uploading all assemblies and MAGs to the European Nucleotide Archive for an alternative fast and stable path to data retrieval.
Thomas S B Schmidt, Anthony Fullam, Pamela Ferretti, Askarbek Orakov, Oleksandr M Maistrenko, Hans-Joachim Ruscheweyh, Ivica Letunic, Yiqian Duan, Thea Van Rossum, Shinichi Sunagawa, Daniel R Mende, Robert D Finn, Michael Kuhn, Luis Pedro Coelho, Peer Bork.
SPIRE: a Searchable, Planetary-scale mIcrobiome REsource, Nucleic Acids Research, 2023;, gkad943,
No, our focus is on (shotgun) metagenomes. If you are interested in exploring 16S/18S amplicon data, check out resources such as the Microbe Atlas Project or MGnify.
SPIRE is used by various teams to address a multitude of different questions. For a taxonomic clade of interest, SPIRE may provide a rich complement of metagenome-assembled genomes from various sources to increase diversity for comparative genomics analyses, but also a list of samples and environments (with further metadata) where said clade has been observed. SPIRE provides large pre-computed gene and genome sets ('catalogues') for environments of interest and allows you to "build your own dataset" by combining and subsetting samples and studies. The raw assemblies per sample can likewise be sourced for further analyses beyond genome-centric approaches. Finally, SPIRE provide the means to taxonomically profile metagenomes against its set of over 100k species-level clusters using a custom mOTU database and precomputed taxonomic profiles for all samples in SPIRE will be released in due course.

Methods
megahit/1.2.9 with default parameters.seqtk seq -L 1000 bwa index assembly.fa && bwa mem -v 2 -t 8 assembly.filtered.fasta raw_reads.fq.gz | samtools view -hb -u -F 4 - | samtools sort -m 16G -@4 > reads_aligned_to_assembly.bamjgi_summarize_bam_contig_depths --outputDepth reads_aligned_to_assembly.depths reads_aligned_to_assembly.bammetabat2 --verbose -i assembly.filtered.fasta -a reads_aligned_to_assembly.depths -t 4 --seed 1987 -o output.bins)prodigal/2.6.3-2-gfe80417 with parameter -p meta.